
Rejoignez-nous le 30 avril : dévoilement de Parasoft C/C++test CT pour l'excellence en matière de tests continus et de conformité | En savoir plus
Aller à la section
Parcourir les directives de validation de la FDA pour la cybersécurité des dispositifs médicaux

13 novembre 2023
13 min lire
Avec l'ajout par la FDA d'exigences supplémentaires à leur validation et à leurs normes de cybersécurité, les fabricants de logiciels médicaux doivent adopter une analyse statique pour garantir que leurs logiciels répondent à ces nouvelles normes de sécurité. Lisez la suite pour savoir comment mettre en œuvre l'analyse statique pour répondre à ces exigences de sécurité.
Aller à la section
Aller à la section
Alors que la FDA ajoute davantage d'exigences en matière de cybersécurité à ses directives de validation de logiciels, les fabricants de dispositifs médicaux peuvent se tourner vers l'analyse statique, la méthode la plus efficace pour répondre aux problèmes de sûreté et de sécurité et fournir des logiciels prévisibles.
Les fabricants de dispositifs médicaux continuent de se concentrer sur l’amélioration des processus de développement de logiciels pour deux raisons principales.
- Répondez aux menaces de sécurité croissantes.
- Satisfaire aux exigences de la FDA, qui se précisent.
Comprendre les nouvelles directives de la FDA en matière de cybersécurité
Les nouvelles directives de cybersécurité de la FDA pour les dispositifs médicaux ont été publiées en septembre 2023 et fournissent des recommandations complètes à l'industrie concernant la conception, l'étiquetage et la documentation des dispositifs de cybersécurité à inclure dans les soumissions préalables à la commercialisation des dispositifs présentant un risque de cybersécurité.
Ces lignes directrices visent à aider les fabricants à identifier et à atténuer les risques de cybersécurité tout au long du cycle de vie d'un dispositif médical, depuis la conception et le développement jusqu'à la fabrication, la maintenance et la surveillance après commercialisation. Il fournit également des recommandations pour communiquer les risques de cybersécurité aux prestataires de soins de santé et aux patients.
Les nouvelles lignes directrices s'appuient sur les précédents documents d'orientation de la FDA en matière de cybersécurité, mais elles comprennent également un certain nombre de recommandations nouvelles et mises à jour, telles que :
- Une obligation pour les fabricants de mettre en œuvre un programme complet de gestion des risques de cybersécurité.
- Une obligation pour les fabricants de soumettre une nomenclature logicielle (SBOM) pour leurs appareils.
- Une obligation pour les fabricants d'élaborer et de mettre en œuvre des plans de cybersécurité pour le support après commercialisation.
- Recommandations pour communiquer les risques de cybersécurité aux prestataires de soins de santé et aux patients.
Auparavant, la FDA se concentrait sur sécurité fonctionnelle aspects des systèmes, mais maintenant cybersécurité est un sujet d’égale importance. Même si la sûreté et la sécurité sont similaires – et on pourrait facilement affirmer que les deux concernent la création de logiciels prévisibles – la FDA considère la cybersécurité comme quelque chose qui nécessite une attention et des mesures spécifiques.
L'importance de la validation des processus dans les dispositifs médicaux
La validation d'un processus est la collecte et l'évaluation de données qui établissent la preuve scientifique qu'un processus est capable de fournir systématiquement un produit de qualité. Dans le contexte des dispositifs médicaux, la validation des processus est essentielle pour garantir la sécurité et l'efficacité des dispositifs tout au long de leur cycle de vie.
La validation des processus est particulièrement importante pour les dispositifs médicaux contrôlés par un logiciel, car celui-ci peut être complexe et difficile à tester de manière exhaustive. Par exemple, un dispositif médical contrôlé par logiciel peut comporter des millions de lignes de code, et il n'est pas possible de tester tous les scénarios et combinaisons possibles d'entrées et de sorties sans utiliser les deux. analyse de code statique et analyse dynamique.
Vous trouverez ci-dessous trois raisons principales pour lesquelles la validation des processus est importante.
- Assurance qualité. La validation des processus permet de garantir que les dispositifs médicaux sont fabriqués de manière cohérente et fiable, réduisant ainsi le risque de défauts et garantissant un haut niveau de qualité.
- Sécurité du patient. En validant le processus de fabrication, les risques et défauts potentiels peuvent être identifiés et atténués, améliorant ainsi la sécurité des patients.
- Conformité aux exigences réglementaires. Le respect des exigences réglementaires, telles que la réglementation du système qualité (QSR) de la FDA, nécessite l'utilisation de processus validés. Ne pas le faire peut entraîner des problèmes de conformité.
Exigences réglementaires et document d'orientation de la FDA
Les exigences réglementaires décrites dans le Document d'orientation de la FDA sur la cybersécurité des dispositifs médicaux, sont conçus pour répondre aux préoccupations croissantes concernant la sécurité et la sûreté des dispositifs médicaux dans un environnement de soins de santé de plus en plus connecté et numérique. Voici une explication des exigences réglementaires.
Autorité statutaire
La FDA dispose désormais d'un nouveau pouvoir statutaire en vertu de la Food and Drug Omnibus Reform Act (FDORA), promulguée en décembre 2022. Cette loi permet à la FDA d'exiger des informations sur la cybersécurité dans les soumissions de dispositifs médicaux pour les « cyber-dispositifs » et oblige les fabricants à prendre des mesures spécifiques. des actions pour démontrer une assurance raisonnable que leurs appareils et systèmes associés sont « cybersécurisés ». Le non-respect de ces exigences est considéré comme un acte interdit, passible de poursuites.
Définition de « cyber appareil »
Le terme « cyber-appareil » est nouvellement défini dans la loi comme un appareil qui comprend un logiciel validé, installé ou autorisé par le sponsor, peut se connecter à Internet et contient des caractéristiques technologiques qui pourraient être vulnérables aux menaces de cybersécurité. Cette définition permet de clarifier quels appareils sont soumis aux nouvelles exigences en matière de cybersécurité.
Inclusion d'informations sur la cybersécurité dans les soumissions préalables à la commercialisation
Les nouvelles directives exigent que les fabricants incluent des informations spécifiques sur la cybersécurité dans leurs soumissions préalables à la commercialisation auprès de la FDA. Ces soumissions préalables à la commercialisation peuvent inclure des soumissions d'approbation préalable à la commercialisation (PMA), 510(k) ou de novo. Les informations de cybersécurité sont nécessaires pour garantir que l'appareil répond aux exigences de cybersécurité décrites dans la loi.
Surveillance et résolution des vulnérabilités en matière de cybersécurité après commercialisation
Les fabricants d'appareils sont tenus de soumettre un plan de surveillance, d'identification et de traitement des vulnérabilités et des exploits de cybersécurité après commercialisation. Ce plan doit décrire l'approche du fabricant pour surveiller et gérer en permanence les risques de cybersécurité qui peuvent apparaître après la mise sur le marché de l'appareil.
Assurance raisonnable de la cybersécurité
Les fabricants doivent concevoir, développer et maintenir des processus et des procédures pour fournir une assurance raisonnable que l’appareil et ses systèmes associés sont « cybersécurisés ». Cette exigence souligne la nécessité d'une approche proactive pour intégrer la sécurité dans la conception de l'appareil et la maintenir tout au long de son cycle de vie.
Nomenclature du logiciel
Les fabricants doivent fournir une nomenclature logicielle (SBOM) dans le cadre de leurs soumissions. Ce projet de loi devrait détailler les composants logiciels commerciaux, open source et disponibles dans le commerce utilisés dans l'appareil. Ces informations sont cruciales pour identifier et corriger les vulnérabilités potentielles de la pile logicielle.
Conformité aux exigences supplémentaires de la FDA
Le document d'orientation autorise également la FDA à établir des exigences supplémentaires par le biais de réglementations pour démontrer que l'appareil et les systèmes associés sont cybersécurisés. Les fabricants doivent être prêts à se conformer à toutes les exigences supplémentaires décrites par la FDA.
Acte interdit en cas de non-conformité
Un aspect clé des exigences réglementaires est l’inclusion d’un nouvel acte statutaire interdit. Cela signifie que le non-respect des exigences de cybersécurité de la FDA n'est pas seulement un problème de conformité, mais également une violation de la loi. Le gouvernement a le pouvoir de poursuivre pénalement les violations de ces exigences ou d’intenter des mesures d’injonction contre les entreprises qui ne s’y conforment pas.
Comment les fabricants de dispositifs médicaux peuvent se conformer
Les fabricants de dispositifs médicaux doivent se conformer à de nombreuses exigences pour garantir la sécurité, l'efficacité et la qualité de leurs produits. Nous explorerons les aspects clés de la conformité aux exigences réglementaires de la FDA en matière de dispositifs médicaux, en mettant davantage l'accent sur la résolution des problèmes de sécurité des dispositifs médicaux et sur la conception de contrôles et de protocoles de validation pour les fabricants.
Répondre aux problèmes de sécurité liés aux dispositifs médicaux
Pour se conformer aux réglementations de la FDA et donner la priorité à la sécurité des patients, les fabricants de dispositifs médicaux doivent répondre efficacement aux problèmes de sécurité. Voici quelques conseils pour répondre aux problèmes de sécurité des dispositifs médicaux :
- Standardisez votre code et effectuez une évaluation des risques. La première étape pour disposer d’un dispositif logiciel médical dépourvu de problèmes de sécurité consiste à écrire le logiciel avec des règles de codage standardisées. Pour les logiciels déjà existants, vous pouvez vérifier et nettoyer la base de code pour vous assurer qu'elle est conforme aux normes de codage acceptables. Les fabricants de dispositifs médicaux peuvent exploiter des solutions d'analyse de code statique pour tester et analyser le code source de leurs logiciels. En plus de l'analyse de code statique, d'autres types de tests logiciels peuvent aider à fournir d'excellents logiciels, notamment les tests d'intégration, les tests de sécurité, les tests unitaires, les tests système et les tests d'utilisabilité. Nettoyer la base de code du logiciel et effectuer une analyse des risques aide les fabricants à identifier les failles de sécurité potentielles de leurs appareils. Cela inclut l’évaluation des risques liés aux violations de données, aux accès non autorisés et à l’intégrité des fonctionnalités de l’appareil.
- Mises à jour logicielles et gestion des correctifs. Mettez régulièrement à jour et corrigez les logiciels de l’appareil pour remédier aux vulnérabilités connues. Les mises à jour en temps opportun aident à prévenir les failles de sécurité susceptibles d'exploiter des logiciels obsolètes.
- Stockage sécurisé des données. Assurez-vous que les données du patient sont stockées en toute sécurité, à la fois sur l'appareil et pendant la transmission des données. Utilisez des pratiques de stockage de données sécurisées et des protocoles de cryptage recommandés par l’industrie pour protéger la vie privée des patients et vous conformer aux réglementations en matière de protection des données.
- Collaborez avec des experts en cybersécurité. Envisagez de collaborer avec des experts en cybersécurité et des professionnels spécialisés dans la sécurité des dispositifs médicaux. Leur expertise peut aider les fabricants à identifier et à atténuer les risques de sécurité potentiels.
Contrôles de conception et protocole de validation pour les fabricants
Des contrôles de conception et des protocoles de validation appropriés sont des éléments essentiels pour garantir que les dispositifs médicaux répondent aux exigences réglementaires de la FDA. Ces contrôles aident les fabricants à développer des dispositifs sûrs et efficaces qui offrent les avantages escomptés aux patients.
Voici les principales considérations pour la mise en œuvre de contrôles de conception et de protocoles de validation.
- Planification de la conception et du développement. Les fabricants doivent établir un plan de conception et de développement clair qui décrit la portée, les objectifs et les attentes du dispositif médical. Ce plan doit inclure la gestion des risques, des études d’utilisabilité et la conformité réglementaire.
- Concevoir les entrées et les sorties. Définissez les entrées de conception qui spécifient les exigences du périphérique et les sorties de conception qui décrivent les caractéristiques du périphérique. Assurez-vous que les entrées de conception correspondent aux besoins des utilisateurs et aux exigences réglementaires.
- Verification ET VALIDATION. Mettez en œuvre des processus rigoureux de vérification et de validation pour confirmer que l’appareil répond à ses exigences de conception et à son utilisation prévue. Cela comprend le test et l'évaluation des performances et de la sécurité de l'appareil.
- Fichier d'historique de conception (DHF). Maintenir un DHF complet qui enregistre toutes les activités de conception et de développement, y compris la documentation des contrôles de conception, les résultats des tests et les modifications de conception.
- Soumissions réglementaires. La soumission de notifications préalables à la commercialisation (510(k)) précises et complètes, de demandes De Novo ou de demandes d'approbation préalable à la commercialisation (PMA) est essentielle pour obtenir l'autorisation ou l'approbation de la FDA pour vos dispositifs médicaux. Ces soumissions fournissent à la FDA les informations nécessaires sur la sécurité et l'efficacité de votre appareil, les aidant ainsi à évaluer son adéquation au marché. Le fait de ne pas remplir correctement ces soumissions peut entraîner des retards, voire le rejet de votre produit.
- Surveillance post-commercialisation (PMS). La mise en place d'un système PMS est essentielle pour surveiller et signaler les événements indésirables, les plaintes et les dysfonctionnements des appareils. Le respect des exigences du PMS permet d'identifier les problèmes de sécurité et de mettre en œuvre rapidement des actions correctives. Il garantit que les fabricants surveillent activement les performances de leurs appareils et résolvent tout problème résultant de leur utilisation.
Outils et méthodes pour la conformité FDA
Les fabricants peuvent utiliser divers outils et méthodes pour atteindre et maintenir la conformité aux réglementations de la FDA relatives aux dispositifs médicaux. Les outils et méthodes essentiels qui peuvent contribuer à la conformité à la FDA sont présentés ci-dessous.
Analyse statique pour répondre aux exigences de la FDA
L'analyse statique joue un rôle essentiel en aidant les fabricants de dispositifs médicaux à répondre aux exigences strictes établies par la FDA.
Voici 5 raisons d’adopter l’analyse statique pour répondre aux exigences de la FDA.
- Détection précoce des problèmes. Outils d'analyse statique peut identifier les problèmes potentiels tels que les erreurs de codage, les vulnérabilités de sécurité et les défauts de conception à un stade précoce du processus de développement. Cette détection précoce permet d'apporter des corrections en temps opportun, réduisant ainsi le risque de modifications coûteuses et fastidieuses plus tard dans le cycle de développement. Les normes de codage recommandées pour C et C++ sont MISRA C / C ++ et CERT C/C++.
- Qualité logicielle améliorée. Un logiciel de haute qualité est impératif pour la sécurité et l’efficacité des dispositifs médicaux. L'analyse statique aide les développeurs à créer un code plus propre, plus fiable et plus sécurisé. Cela facilite non seulement la conformité avec la FDA, mais atténue également le risque de problèmes post-commercialisation et de problèmes de sécurité.
- Conformité en matière de cybersécurité. Compte tenu de l’importance croissante de la cybersécurité pour les dispositifs médicaux, la FDA y accorde une grande importance. Les outils d'analyse statique peuvent identifier les faiblesses et les vulnérabilités de sécurité du logiciel, garantissant ainsi l'alignement avec les directives de la FDA en matière de cybersécurité.
- Documentation réglementaire. L'analyse statique peut fournir une documentation et des rapports complets qui démontrent l'examen approfondi du logiciel et les mesures prises pour résoudre les problèmes identifiés. Cette documentation est essentielle pour les soumissions et les inspections de la FDA.
- Validation simplifiée. En identifiant et en traitant rapidement les problèmes potentiels, l’analyse statique peut accélérer le processus de validation, qui constitue une étape importante dans la conformité FDA. Cela permet de réduire les délais de mise sur le marché des dispositifs médicaux.
Comment assurer une bonne gestion des risques
Compte tenu de la place de la gestion des risques dans la réalisation des objectifs ou des exigences globales de la FDA, examinons comment vous pouvez parvenir à une gestion des risques appropriée.
- L'évaluation des risques. L'évaluation des risques dans la fabrication de dispositifs médicaux est la première étape pour identifier et comprendre les dangers potentiels associés à un dispositif médical. Son objectif est d'évaluer systématiquement la conception du dispositif, son utilisation prévue, sa biocompatibilité et d'autres facteurs de risque afin d'identifier les risques potentiels. Un exercice d’évaluation des risques implique généralement la formation d’une équipe interfonctionnelle dotée d’expertises diverses, comprenant des ingénieurs, des cliniciens et des spécialistes de la réglementation.
- Analyse de risque. L'analyse des risques vise à évaluer et hiérarchiser les risques identifiés en attribuant un score de risque basé sur la probabilité d'occurrence et la gravité du préjudice potentiel. L'analyse des risques consiste à quantifier chaque risque en évaluant sa probabilité et son impact potentiel. Selon votre entreprise, une matrice des risques ou un autre système de notation peut être utilisé pour hiérarchiser les risques. Les éléments à haut risque sont prioritaires pour des mesures ultérieures.
- Atténuation des risques. L'atténuation des risques implique l'élaboration de stratégies visant à réduire ou à contrôler les risques identifiés à un niveau acceptable. Il vise à améliorer la sécurité et l’efficacité du dispositif médical. Les stratégies d'atténuation peuvent prendre diverses formes, notamment des modifications de conception, des dispositifs de sécurité, de meilleurs matériaux, une formation améliorée des utilisateurs ou des instructions d'utilisation améliorées. L’objectif est de répondre aux risques identifiés de manière à minimiser leur impact.
- Documentation. Une documentation appropriée est essentielle pour la transparence, la responsabilité et la conformité réglementaire. Il fournit un enregistrement du processus de gestion des risques et aide à démontrer la diligence raisonnable auprès des autorités réglementaires. Tous les aspects du processus de gestion des risques, y compris les évaluations des risques, les analyses et les plans d'atténuation, doivent être méticuleusement documentés. Cette documentation est conservée tout au long du cycle de vie du produit.
- Amélioration continue. Le processus de gestion des risques doit être un système dynamique et évolutif. L'amélioration continue garantit que vos pratiques de gestion des risques restent efficaces pour faire face aux nouveaux risques et aux conditions changeantes du marché. Des examens et des mises à jour réguliers sont effectués pour intégrer les enseignements tirés des projets antérieurs et s'adapter à l'évolution de la technologie et des réglementations. Cela peut impliquer de revoir et de réviser les évaluations des risques, les analyses et les plans d’atténuation.
Comment l'analyse statique pour C/C++ peut aider à la gestion des risques
Beaucoup de nos clients de dispositifs médicaux, en partant de zéro, ont réussi à introduire analyse statique pour C / C ++ en suivant ces étapes:
- Passez en revue les directives existantes dans l’organisation. Même s'ils sont conçus pour être appliqués manuellement, mappez-les autant que possible aux vérificateurs fournis avec l'outil d'analyse statique. Un outil d’analyse statique mature couvrira probablement la plupart d’entre eux. Vous pouvez envisager de créer des vérificateurs personnalisés pour les directives restantes qui n'ont pas pu être mappées immédiatement aux vérificateurs d'analyse statique.
- Passez en revue les normes de codage populaires, en particulier celles créées dans un souci de sécurité. Sélectionnez un sous-ensemble de directives que votre équipe devra suivre. Lors de la sélection des lignes directrices, il est logique de suivre la catégorisation des normes et de sélectionner les lignes directrices classées comme les plus importantes. Par exemple, dans le CERT, vous souhaiterez probablement commencer par les directives L1, et dans MISRA C 2023, vous souhaiterez revoir les directives obligatoires.
- Définissez la configuration de l'outil d'analyse statique et incluez les directives spécifiques à votre organisation et les directives sélectionnées telles que CERT. N'activez pas tous les contrôleurs en même temps. Commencez plutôt par un petit sous-ensemble pour éviter d’inonder les développeurs de violations.
- Assurez-vous que vos développeurs peuvent analyser leur code immédiatement après sa création. Il est également logique d'inclure l'analyse statique dans le processus CI/CD.
- Au fur et à mesure que les développeurs progressent et nettoient le code source, assurez-vous d'activer progressivement davantage de vérificateurs de votre liste. En fin de compte, vous souhaitez fournir la preuve que vous respectez l’ensemble des directives que vous avez sélectionnées et non que vous vous êtes arrêté à mi-chemin. Pour mieux vous orienter dans le processus et comprendre vos progrès actuels, vous pouvez déployer un système de reporting central pour vous aider à regrouper les données de test et à surveiller le travail des développeurs. Voir un exemple ci-dessous :
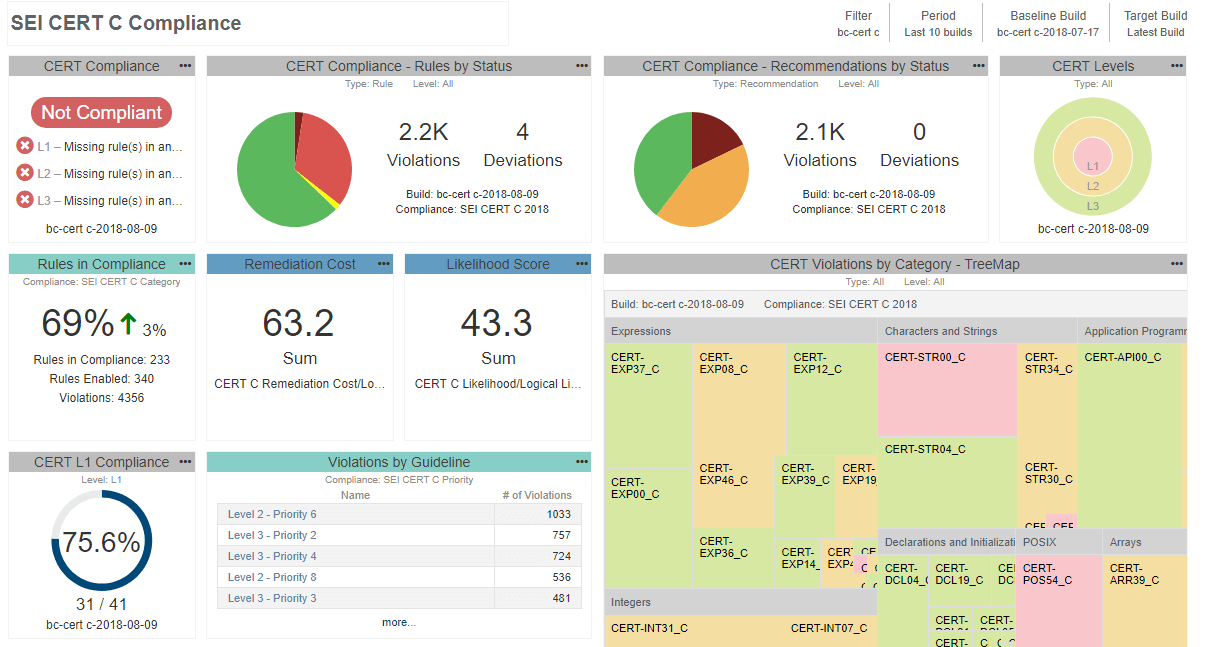
Étant donné que les rapports d’analyse statique font partie du système de gestion de la qualité, vous ne pouvez pas utiliser n’importe quel outil. La FDA exige que tous les outils utilisés pour le développement et la vérification du logiciel soient validés pour l'utilisation prévue. Il existe différentes manières de démontrer l'adéquation de l'outil à une utilisation dans le cadre d'un développement critique pour la sécurité. Selon le risque présenté par le dispositif, cela peut être aussi simple que de réutiliser un certificat de conformité ou de suivre le processus plus long de qualification d'outils.
Utilisation d'une certification TÜV SÜD
Pour l'utilisateur final, l'option la plus pratique est de s'attribuer le mérite du travail effectué par le fournisseur de l'outil et de réutiliser la certification accordée pour l'outil de test par un organisme de certification externe tel que TÜV SÜD. Parasoft C / C ++test , par exemple, est recouvert de une certification TÜV SÜD qui peut être réutilisé pour démontrer son aptitude au développement de logiciels conformément aux normes médicales telles que la CEI 62304.
Exécution de la qualification des outils
Pour les appareils à haut risque tels que la classe C, vous devrez peut-être valider l'outil en interne dans votre environnement de développement. L'intention est de fournir la preuve que l'outil fonctionne conformément à ses exigences opérationnelles, recueillies dans l'environnement de développement du projet. Il s'agit d'un processus très fastidieux et long.
La meilleure situation est si votre fournisseur d'outils peut vous soutenir dans cet effort et vous fournir un kit de qualification d'outils spécial contenant des cas de test bien conçus, ainsi que le cadre d'automatisation pour les exécuter dans l'environnement de développement du projet, et générer automatiquement la documentation qui peut servir de la preuve pour la validation de l’outil. Là encore, le produit phare de Parasoft, C/C++test, fournit un kit de qualification d'outils automatisé.
Études de cas et applications concrètes
Nous disposons d'une multitude d'études de cas d'applications réelles des outils d'analyse statique Parasoft. Mais examinons deux réussites distinctes qui illustrent comment notre solution d'analyse statique a joué un rôle important en aidant les fabricants de dispositifs médicaux à surmonter leurs défis uniques et à répondre aux exigences de la FDA.
Témoignages de réussite de fabricants de dispositifs médicaux
Les études de cas suivantes illustrent comment les solutions de tests logiciels de Parasoft ont joué un rôle déterminant pour aider les fabricants de dispositifs médicaux à relever leurs défis uniques et à atteindre leurs objectifs. Inovytec et Smiths Medical ont tous deux constaté des améliorations remarquables en termes de qualité du code, de conformité et d'efficacité des tests grâce à leur collaboration avec Parasoft.
Le parcours d'Inovytec vers la certification FDA 510(k)
Inovytec, une entreprise dédiée à la production de dispositifs médicaux, s'est lancée dans une mission visant à obtenir la certification FDA 510(k) pour son ventilateur Ventway Sparrow. Leur défi consistait à fournir un code propre tout en respectant les réglementations de la FDA. La solution d'analyse de code statique C/C++ de Parasoft est venue à leur secours.
L'équipe de développement logiciel d'Inovytec a personnalisé le test Parasoft C/C++ pour s'aligner sur les exigences strictes de la FDA. Chaque fois qu'ils se préparaient à publier une nouvelle version du logiciel, ils s'assuraient que l'analyse statique de Parasoft était configurée pour s'exécuter conformément aux définitions de la réglementation FDA. Le résultat a été non seulement une amélioration de la qualité du code, mais également un succès retentissant dans la réussite de 100 % des règles et directives de certification FDA 510(k). Parasoft est devenu la solution de test préférée chez Inovytec, et leur collaboration avec ESL, un distributeur de produits Parasoft en Israël, a fourni un soutien et une expertise essentiels en cas de besoin.
Adoption par Smiths Medical du développement piloté par les tests (TDD)
Smiths Medical, fabricant renommé de dispositifs médicaux spécialisés, a rencontré une série de défis dans sa quête pour développer des produits de haute qualité et critiques pour la sécurité. logiciel de dispositif médical. Les tests automatisés jouent un rôle crucial dans la stratégie de test de Smiths Medical.
Les efforts antérieurs visant à intégrer des outils n’ont pas abouti. L'équipe de développement recherchait une solution capable d'améliorer l'ensemble de son processus de test en adoptant une nouvelle perspective centrée sur les tests unitaires et le développement piloté par les tests (TDD), une méthodologie qui intègre la conception, les tests et le développement de code. Ils avaient besoin d'un outil capable de s'intégrer dans leur pipeline de tests et d'améliorer leur culture globale de développement. Le test Parasoft C/C++ s'est avéré être la réponse à leurs défis.
L'équipe logicielle a non seulement réussi à adopter le TDD, mais a également bénéficié d'une meilleure stabilité des tests, d'une couverture de code améliorée et d'un processus de qualification d'outils rationalisé, ce qui était essentiel pour les applications critiques pour la sécurité. Avec Parasoft, Smiths Medical a pu transformer ses processus de développement, en faisant des tests une partie intégrante de son pipeline de logiciels et, en fin de compte, en garantissant la livraison de dispositifs médicaux sûrs et de haute qualité.
Surmonter les défis de la validation FDA
Le processus de validation FDA peut être complexe et difficile pour les fabricants de dispositifs médicaux. Cependant, les fabricants peuvent prendre un certain nombre de mesures pour surmonter ces défis.
- Commencer de bonne heure. Le processus de validation de la FDA doit commencer dès le début du cycle de développement du produit. Cela donnera aux fabricants plus de temps pour planifier et exécuter le processus de validation, et cela aidera à identifier et à résoudre tout problème potentiel dès le début.
- Utilisez une approche basée sur les risques. Le processus de validation de la FDA doit être basé sur une évaluation des risques du dispositif. Les fabricants doivent concentrer leurs efforts de validation sur les composants et fonctions les plus critiques de l’appareil.
- Faire appel à du personnel qualifié. Le processus de validation de la FDA doit être mené par du personnel qualifié ayant l'expérience des réglementations de la FDA et des meilleures pratiques de validation.
- Documentez le processus de validation. Les fabricants doivent documenter minutieusement le processus de validation de la FDA. Cela aidera les fabricants à démontrer à la FDA qu'ils ont validé leur dispositif conformément aux réglementations de la FDA.
Prochaines étapes pour naviguer dans les directives de validation de la FDA
La FDA fournit des conseils de validation essentiels aux fabricants de dispositifs médicaux afin de garantir la qualité des produits et la sécurité des patients. À mesure que les réglementations et les directives évoluent, il est crucial de rester au courant de ces changements et de planifier leur conformité future. Les fabricants de dispositifs médicaux peuvent y parvenir de deux manières : se préparer aux futures directives de la FDA et rester en conformité.
Se préparer aux futures directives de la FDA
La FDA met constamment à jour et révise ses documents d’orientation destinés aux fabricants de dispositifs médicaux. Pour se préparer aux futures directives de la FDA, les fabricants de dispositifs médicaux doivent :
- Surveillez le site Web de la FDA pour connaître les mises à jour des documents d'orientation. La FDA publie des mises à jour de ses documents d'orientation sur son site Web. Les fabricants doivent consulter régulièrement le site Web de la FDA pour connaître les mises à jour des documents d'orientation pertinents pour leurs appareils.
- Participez aux ateliers et webinaires de la FDA. La FDA organise fréquemment des ateliers et des webinaires sur divers sujets liés à la réglementation des dispositifs médicaux. Les fabricants devraient participer à ces événements pour se tenir au courant des dernières réflexions de la FDA sur la réglementation des dispositifs médicaux.
- Réseautez avec d’autres fabricants de dispositifs médicaux. Les fabricants peuvent réseauter avec d’autres fabricants de dispositifs médicaux pour partager des informations et les meilleures pratiques afin de se conformer aux réglementations de la FDA.
Assurer une conformité continue
Les fabricants de dispositifs médicaux doivent garantir une conformité continue aux réglementations de la FDA. Pour ce faire, les fabricants doivent :
- Mettre en place et maintenir un système qualité. Un système qualité est un ensemble de procédures et de processus que les fabricants utilisent pour garantir la sécurité et l'efficacité de leurs appareils. Les fabricants doivent établir et maintenir un système qualité qui répond aux exigences du règlement sur le système qualité de la FDA.
- Réaliser des audits réguliers de leur système qualité. Les fabricants doivent effectuer des audits réguliers de leur système qualité pour identifier et traiter tout domaine de non-conformité.
- Soumettez les rapports MDR à la FDA. Les fabricants sont tenus de soumettre des rapports sur les dispositifs médicaux (MDR) à la FDA pour les événements indésirables survenus avec leurs dispositifs. En soumettant des rapports MDR à la FDA, les fabricants peuvent aider la FDA à identifier et à résoudre les problèmes de sécurité liés à leurs appareils.
Au fil du temps, la mise en conformité avec la FDA s'est avérée être une tâche très rigoureuse et longue. Cependant, grâce aux outils d’analyse statique et d’analyse dynamique, ces défis peuvent être surmontés. Les outils d'analyse statique offrent des fonctionnalités avancées d'analyse de code statique, qui permettent d'identifier et de corriger les problèmes liés aux logiciels et au code dès le début du processus de développement.
L’introduction de l’analyse statique est un effort dédié, qui nécessite du temps et de l’argent pour les développeurs. Mais c'est un moyen éprouvé de renforcer votre système contre les attaques malveillantes. Le déploiement d'une analyse statique avec un ensemble bien pensé de directives de sécurité vous permet de créer des systèmes capables de résister à de futures attaques imprévues.
Conformité logicielle CEI 62304 dans l'industrie médicale
« MISRA », « MISRA C » et le logo triangulaire sont des marques déposées de The MISRA Consortium Limited. ©The MISRA Consortium Limited, 2021. Tous droits réservés.



